
Hyperphenylalaninemia (HPA) is a metabolic disorder characterized by elevated levels of the blood’s amino acid phenylalanine (Phe), which can have significant health implications if left untreated.
While primarily associated with phenylketonuria (PKU), HPA can also arise from deficiencies in tetrahydrobiopterin (BH4), a cofactor for several enzymes, including phenylalanine hydroxylase (PAH)1,2. Understanding these conditions is crucial for those affected, as early detection and management can lead to better health outcomes.
This blog post will explain in detail what BH4 is and its roles. It will explore the genetic mutations affecting BH4 metabolism. It will examine the implications of BH4 deficiency. Finally, it will discuss how to distinguish between PKU and BH4 deficiencies.
What is BH4?
Tetrahydrobiopterin (BH4) is a naturally occurring cofactor that is critical in various enzymatic reactions within the body3. Cofactors are non-protein chemical compounds or metallic ions essential for enzyme activity. They act as “helper molecules,” assisting enzymes in catalyzing biochemical reactions effectively.
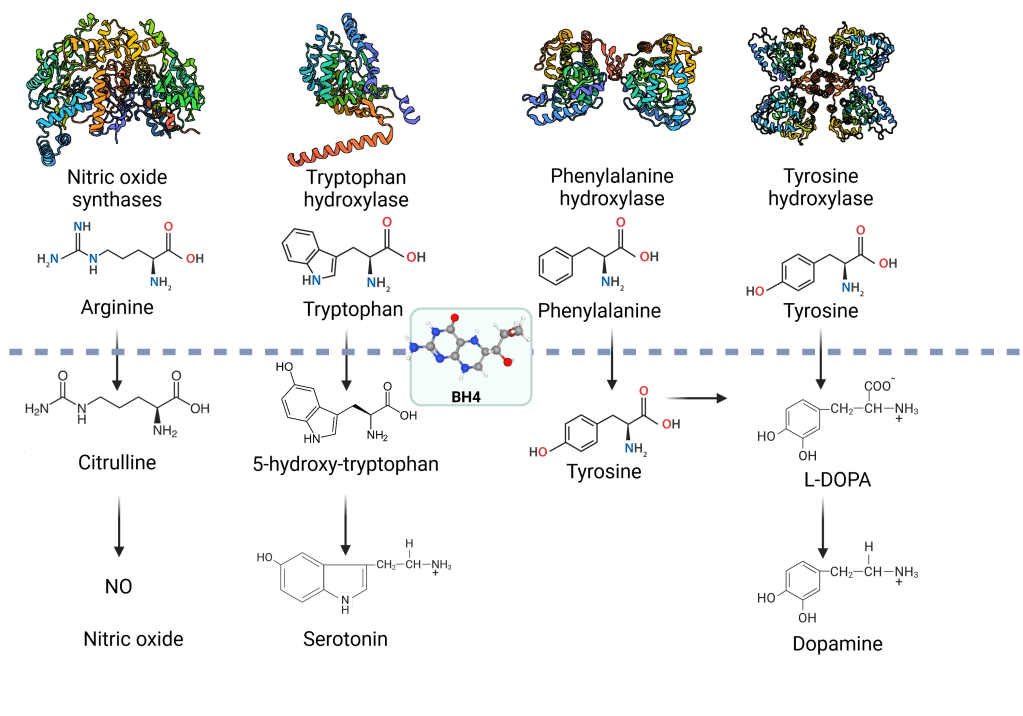
BH4 is particularly important in the metabolism of the amino acid phenylalanine (Phe). It is a cofactor for the phenylalanine hydroxylase (PAH), which converts Phe into tyrosine. Tyrosine is another amino acid vital for synthesising neurotransmitters, making BH4 essential for proper brain function and overall metabolic processes4,5.
Role of BH4 as a Cofactor
BH4 enhances or enables the activity of enzymes. In the case of PAH, BH4 stabilizes the enzyme and facilitates its catalytic function. Without adequate levels of BH4, PAH cannot effectively convert Phe to tyrosine. This leads to elevated levels of Phe in the blood. This condition is known as hyperphenylalaninemia (HPA).
Beyond PAH, BH4 is also a cofactor for other important enzymes, including:
- Tyrosine hydroxylase (TH): Converts tyrosine to L-DOPA, a precursor for dopamine.
- Tryptophan hydroxylase (TPH): Converts tryptophan to serotonin.
- Nitric oxide synthases (NOS): Catalyze nitric oxide production from L-arginine6.
Other roles of BH4 have been uncovered
Latini et al. research group has revealed new significant roles for BH4 metabolism.
The group described the physiological properties of the BH4 pathway. Its functions include acting as an antioxidant and anti-inflammatory agent. It also serves as a mitochondrial activator and memory enhancer in the nervous system.
They have also demonstrated the impact of genetic or pharmacological inactivation of Gch1 on T cell proliferation and tissue infiltration. Additionally, the group has shown that excessive activation of the BH4 pathway can lead to pathogenic effects. These effects include mitochondrial dysfunction, increased immune system aggressiveness, and worsening of symptoms in chronic diseases.
Furthermore, the potential for restoring the physiological, metabolic flow of the BH4 pathway was highlighted. This can alleviate or normalize pain clinical scores. It offers new therapeutic possibilities for treating chronic inflammatory conditions5–7.
Effects of Various Mutations on BH4
Mutations in several genes can lead to BH4 deficiencies, each affecting the biosynthesis or recycling of this essential cofactor:
- GCH1: This gene encodes GTP cyclohydrolase I, the first enzyme in the BH4 biosynthetic pathway. Mutations here can lead to a complete or partial deficiency of BH4, resulting in elevated Phe levels and neurotransmitter deficiencies.
- QDPR: This gene encodes dihydropteridine reductase, responsible for regenerating BH4 from its oxidized form. Mutations can lead to reduced levels of BH4, causing symptoms similar to those seen in GCH1 mutations.
- PCBD1: This gene encodes a protein involved in the recycling of BH4. Defects in this gene can disrupt normal BH4 metabolism, leading to increased Phe levels and neurological symptoms.
- PTS: The PTS gene encodes sepiapterin reductase, another enzyme involved in BH4 synthesis. Mutations here can also result in elevated Phe levels due to insufficient BH4 production 5,8.
Individuals with BH4 deficiencies can have high Phe levels like those observed in classical PKU. The severity of symptoms often depends on the specific mutation and its impact on enzyme function. Besides that, since BH4 is crucial for synthesizing neurotransmitters like dopamine and serotonin, its deficiency can lead to neurological symptoms. Developmental delays can also occur, even with low protein dietary management. Correctly diagnosing BH4 deficiencies is vital as these disorders require different treatment approaches than classical PKU.
Distinguishing PKU from BH4 Deficiencies
Further testing is essential to differentiate between PKU and BH4 deficiencies after newborn screening detects elevated Phe levels. One key diagnostic tool is the BH4 loading test. In this test:
- Infants suspected of having a BH4 deficiency are administered a dose of BH4 (sapropterin dihydrochloride).
- In patients with PKU, the drop in Phe levels after administration is typically minimal to moderate. This indicates that the PAH enzyme has limited residual activity. This condition is termed BH4-responsive PKU.
- In contrast, patients with BH4 deficiencies often show a significant decrease in Phe levels after the loading test. This is due to their impaired ability to synthesize or recycle BH4 9,10.
This differentiation guides treatment approaches and prevents misdiagnosis.
Conclusion
Tetrahydrobiopterin (BH4) is an essential cofactor for several enzymes involved in amino acid metabolism. Its deficiency can lead to elevated phenylalanine levels and significant neurological complications.
Correctly diagnosing whether a patient has classic PKU or a BH4 deficiency is critical for effective treatment and management. Healthcare professionals should be vigilant about potential misdiagnoses and utilize appropriate testing methods to ensure accurate diagnosis and optimal patient care.
Make a one-time donation
Make a monthly donation
Make a yearly donation
Choose an amount
Or enter a custom amount
What If Your Donation Could Change Everything?
Let’s be honest: most people scroll past donation buttons. But you’re not most people.
You’re here because you care about knowledge, about real stories, about making a difference for people who need it most. You know that every breakthrough, every recipe, every insight on raremetabolicinsights.com is a lifeline for someone out there.
Here’s the truth:
This site isn’t powered by big sponsors or faceless corporations. It’s powered by people like you—people who believe that sharing knowledge can change lives.
Why Donate?
Every euro you give is a vote for more content, more recipes, more hope.
Your support means we can publish more often, dive deeper, and reach more families who need answers.
You’re not just donating—you’re joining a movement that refuses to let rare conditions mean rare support.
Imagine This:
Tomorrow, someone finds a recipe here that finally makes their child’s diet easier. Next week, a parent reads a guide that gives them hope. That’s the impact you can have—right now.
If you’ve ever found value here, pay it forward.
Your donation—no matter the size—keeps this community alive and growing.
“The only thing standing between someone and the life-changing information they need… is whether we show up for each other.”
What If Your Donation Could Change Everything?
Let’s be honest: most people scroll past donation buttons. But you’re not most people.
You’re here because you care about knowledge, about real stories, about making a difference for people who need it most. You know that every breakthrough, every recipe, every insight on raremetabolicinsights.com is a lifeline for someone out there.
Here’s the truth:
This site isn’t powered by big sponsors or faceless corporations. It’s powered by people like you—people who believe that sharing knowledge can change lives.
Why Donate?
Every euro you give is a vote for more content, more recipes, more hope.
Your support means we can publish more often, dive deeper, and reach more families who need answers.
You’re not just donating—you’re joining a movement that refuses to let rare conditions mean rare support.
Imagine This:
Tomorrow, someone finds a recipe here that finally makes their child’s diet easier. Next week, a parent reads a guide that gives them hope. That’s the impact you can have—right now.
If you’ve ever found value here, pay it forward.
Your donation—no matter the size—keeps this community alive and growing.
“The only thing standing between someone and the life-changing information they need… is whether we show up for each other.”
What If Your Donation Could Change Everything?
Let’s be honest: most people scroll past donation buttons. But you’re not most people.
You’re here because you care about knowledge, about real stories, about making a difference for people who need it most. You know that every breakthrough, every recipe, every insight on raremetabolicinsights.com is a lifeline for someone out there.
Here’s the truth:
This site isn’t powered by big sponsors or faceless corporations. It’s powered by people like you—people who believe that sharing knowledge can change lives.
Why Donate?
Every euro you give is a vote for more content, more recipes, more hope.
Your support means we can publish more often, dive deeper, and reach more families who need answers.
You’re not just donating—you’re joining a movement that refuses to let rare conditions mean rare support.
Imagine This:
Tomorrow, someone finds a recipe here that finally makes their child’s diet easier. Next week, a parent reads a guide that gives them hope. That’s the impact you can have—right now.
If you’ve ever found value here, pay it forward.
Your donation—no matter the size—keeps this community alive and growing.
“The only thing standing between someone and the life-changing information they need… is whether we show up for each other.”
References
1. Tendi, E. A., Guarnaccia, M., Morello, G. & Cavallaro, S. The Utility of Genomic Testing for Hyperphenylalaninemia. Journal of Clinical Medicine 11, 1061 (2022).
2. Tetrahydrobiopterin Deficiency – Symptoms, Causes, Treatment | NORD. https://rarediseases.org/rare-diseases/tetrahydrobiopterin-deficiency/.
3. Bozaci, A. E. et al. Tetrahydrobiopterin deficiencies: Lesson from clinical experience. JIMD Rep 59, 42–51 (2021).
4. Thöny, B., Ding, Z. & Martínez, A. Tetrahydrobiopterin protects phenylalanine hydroxylase activity in vivo: implications for tetrahydrobiopterin-responsive hyperphenylalaninemia. FEBS Lett 577, 507–511 (2004).
5. Eichwald, T. et al. Tetrahydrobiopterin: Beyond Its Traditional Role as a Cofactor. Antioxidants 12, 1037 (2023).
6. Latini, A. et al. Tetrahydrobiopterin improves hippocampal nitric oxide-linked long-term memory. Molecular Genetics and Metabolism 125, 104–111 (2018).
7. Espíndola, G., Scheffer, D. da L. & Latini, A. Commentary: Urinary Neopterin, a New Marker of the Neuroinflammatory Status in Amyotrophic Lateral Sclerosis. Front. Neurosci. 15, (2021).
8. Fanet, H., Capuron, L., Castanon, N., Calon, F. & Vancassel, S. Tetrahydrobioterin (BH4) Pathway: From Metabolism to Neuropsychiatry. Curr Neuropharmacol 19, 591–609 (2021).
9. Fiege, B. et al. Extended tetrahydrobiopterin loading test in the diagnosis of cofactor-responsive phenylketonuria: a pilot study. Mol Genet Metab 86 Suppl 1, S91-95 (2005).
10. Anjema, K. et al. The neonatal tetrahydrobiopterin loading test in phenylketonuria: what is the predictive value? Orphanet Journal of Rare Diseases 11, 10 (2016).

Leave a comment